about Neurofibromatosis Type 1
Neurofibromatosis type 1 occurs in 1 in 3,000 to 4,000 people worldwide.
References (14 links)
There is no cure or treatment for NF1. Some tumors may be removed, but others, such as plexiforms may be difficult to manage and may only be “debulked”. Optic gliomas may be treated with chemotherapy, though it is only warranted if they progress and cause complications such as vision changes. Bony abnormalities may be treated surgically or through instrumentation.
It is EXTREMELY CRITICAL that people with NF1 are treated by physicians that have a lot of experience in the disorder, especially if there are ANY complications. If you cannot get to an “NF Specialist”, please contact us for other options.
Most of the time, NF1 is diagnosed by its physical symptoms (tumors or café- au-lait spots), or by a family history of the disorder. The café- au-lait spots usually appear within the first two years of a child’s life. A person may be diagnosed with NF1 if they have at least two of the following features:
NF1 may also be diagnosed by sequencing a person’s NF1 gene to identify mutations. But because of the gene’s large size and the high number of possible mutations that can occur, genetic testing is usually not practical – and very expensive.
Doctors may not recommend genetic testing because a diagnosis of NF1 is fairly easy to confirm on the basis of its physical signs, and currently genetic testing offers no extra benefits for treatment. This is something that may be changing.
It is, however, easier to identify a mutation if there is a family history of NF1. If another family member has been tested, and the mutation has been identified, then it becomes relatively easy to look for that same mutation in other family members. Finding the mutation would then confirm the diagnosis of NF1.
Approximately half (50%) of all people with neurofibromatosis type 1 inherit the disorder. It is inherited in an autosomal dominant pattern, which means that only one copy of the defective gene has to be inherited for a baby to be born with NF1. Therefore, each child of a parent with NF1 runs a 50 percent risk of getting the disorder.
In the other half of NF1 cases, a person will have no family history of the disease. The mutation is new and has likely occurred early in life (during the development of the embryo). New mutations are frequently the cause of NF1 because the NF1 gene is very large, making it more likely to have a mutation.
Scientists are still trying to discover why the symptoms vary so much, even among people from the same family. One theory is that each patient’s unique genetic makeup influences the severity of his or her symptoms; meaning that genes other than the NF1 gene might play a role. Scientists call these “modifier genes,” and they could be any of the thousands of genes in the human genome.
How do modifier genes work? Proteins encoded by modifier genes might work in the same biological pathways as neurofibromin, and therefore affect how well these pathways work. Variations in the DNA sequence of a modifier gene may alter its function enough to influence the severity of NF1 in an individual.
To date, scientists have not found any strong candidate NF1 modifier genes.
It’s also possible that environmental events may cause the variability among different NF patients. Also, the different types of mutations that occur in the NF1 gene may be a factor. Currently, over 500 different types of mutations have been discovered in the NF1 gene.
NF type 1 is caused by a mutation in a gene on chromosome 17. This gene provides the instructions for making a protein called neurofibromin. This protein is produced in many cells, including nerve cells and specialized cells surrounding nerves (oligodendrocytes and Schwann cells). Neurofibromin acts as a tumor suppressor, which means that it keeps cells from growing and dividing too rapidly or in an uncontrolled way. Specifically, neurofibromin regulates the activity of another protein called RAS, which promotes cell division. When the NF1 gene is mutated, it usually leads to a shortened version of the neurofibromin protein that cannot bind to RAS or regulate its activity. As a result, the RAS protein is more active. Cells are told to begin dividing and never told when to stop, causing the formation of tumors. As a result, tumors such as neurofibromas can form along nerves throughout the body. It’s not yet known how mutations in the NF1 gene lead to the other features of neurofibromatosis type 1, such as café-au-lait spots and learning disabilities.
Read more about the NF1 gene.
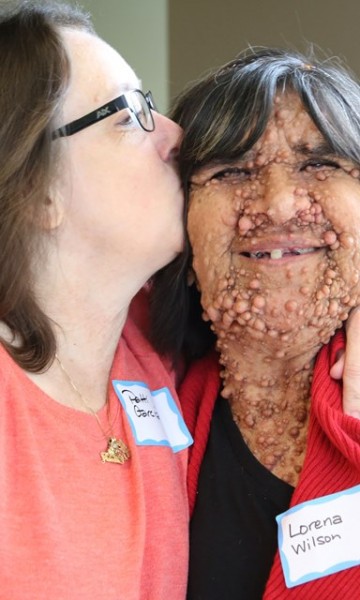
The symptoms and complications of neurofibromatosis type 1 vary widely from patient to patient. However, beginning in early childhood, most people with neurofibromatosis type 1 have 6 or more café-au-lait spots, which are flat patches on the skin that are darker than the surrounding area. Freckles in the underarms and groin typically develop later in childhood.
Most adults with neurofibromatosis type 1 develop neurofibromas, which are noncancerous (benign) tumors that are usually located on or just under the skin. These tumors may also occur in nerves near the spinal cord or along nerves elsewhere in the body. Rarely, these tumors can progress into a malignant tumor. People with neurofibromatosis type 1 also have a slightly increased chance of developing brain tumors, other cancers and leukemia.
Benign growths called Lisch nodules usually appear in the colored part of the eye (the iris) in the teenage or adult years.. Lisch nodules do not interfere with vision. Some affected individuals also develop tumors that grow along the nerve leading from the eye to the brain (the optic nerve). These tumors, which are called optic gliomas, may lead to reduced vision or total vision loss. In some cases, optic gliomas have no effect on vision.
Additional signs and symptoms of neurofibromatosis type 1 include high blood pressure (hypertension), short stature, an unusually large head (macrocephaly), and skeletal abnormalities such as an abnormal curvature of the spine (scoliosis). Although most people with neurofibromatosis type 1 have normal intelligence, learning disabilities and attention deficit hyperactivity disorder (ADHD) occur frequently in affected individuals.
Again, the complications of NF1 may vary WIDELY from individual to individual. Some people may have a few of the complications, some may have many. It is also a chronic disorder that progresses over time. However, for some it may progress very slowly and others it may be more rapid. It also may progress suddenly or a new complication may appear; or it may suddenly slow down. For instance, a tumor may simply stop growing.
There are different ages in which physicians may especially look for different complications.
Neurofibromatosis type 1 (NF1) is a condition characterized by the growth of tumors along nerves in the skin, brain, and other parts of the body. The signs and symptoms of this condition vary widely among affected people.
© 2025 Neurofibromatosis Midwest - non profit 501(c)(t3). Site Map - Privacy Policy
The resources on this site should not be used as a substitute for professional medical care or advice on Neurofibromatosis. Users seeking information about a personal genetic disease, syndrome, or condition should consult with a qualified healthcare professional.





.png)
